A user comment asks the following:
"I wonder how can I alter the size of the "pie" in describe.simmap.... Same question for altering the color...."
Well, there is no way at present to change the color or pie-size in describe.simmap(...,plot=TRUE); however, describe.simmap, it is easy to recreate the posterior probability plot of describe.simmap using the function output - while customizing its attributes.
Here's a quick demo:
> # check package version
> packageVersion("phytools")
[1] ‘0.2.54’
>
> # first simulate some data to use in the demo
> Q<-matrix(c(-2,1,1,1,-2,1,1,1,-2),3,3)
> rownames(Q)<-colnames(Q)<-letters[1:3]
> tree<-sim.history(pbtree(n=50,scale=1),Q)
> x<-tree$states
>
> # now the demo
> mtrees<-make.simmap(tree,x,model="ER",nsim=100)
make.simmap is sampling character histories conditioned on the transition matrix
Q =
a b c
a -2.041867 1.020934 1.020934
b 1.020934 -2.041867 1.020934
c 1.020934 1.020934 -2.041867
(estimated using likelihood);
and (mean) root node prior probabilities
pi =
a b c
0.3333333 0.3333333 0.3333333
Done.
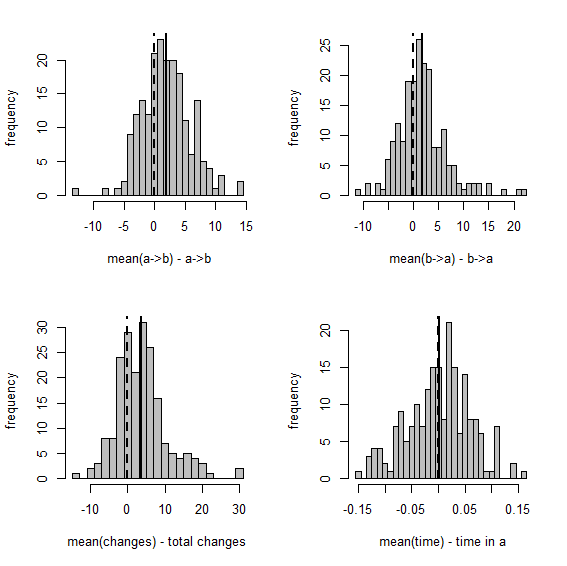
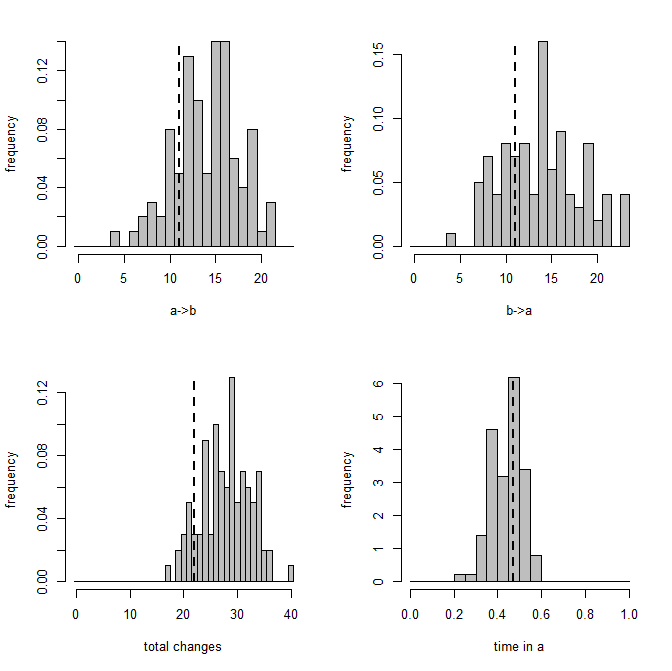
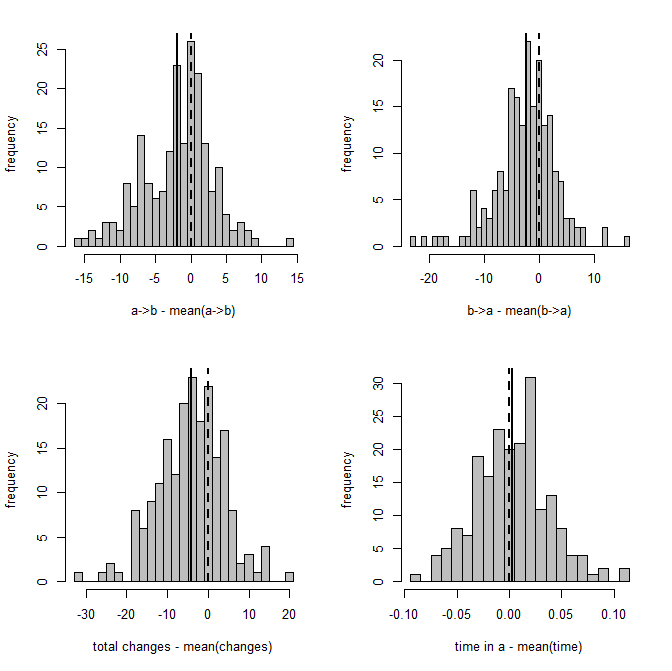
> XX<-describe.simmap(mtrees,plot=FALSE)
100 trees with a mapped discrete character with states:
a, b, c
trees have 23.93 changes between states on average
changes are of the following types:
a,b a,c b,a b,c c,a c,b
x->y 2.11 2.91 4.62 5.77 4.94 3.58
mean total time spent in each state is:
a b c total
raw 2.2816521 4.1930936 4.2593925 10.73414
prop 0.2125603 0.3906316 0.3968081 1.00000
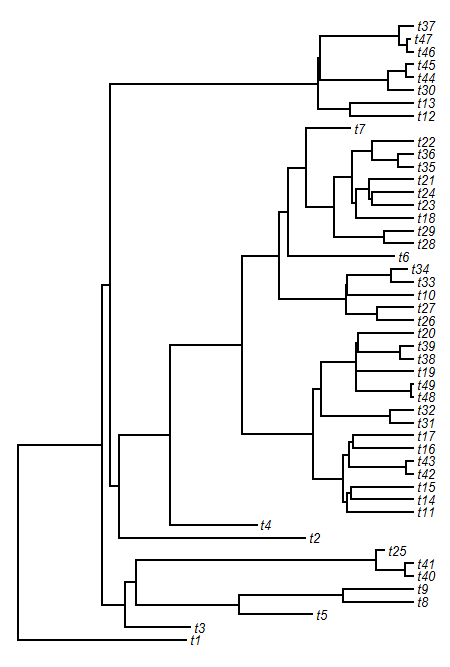
> hh<-max(nodeHeights(tree))*0.02 # for label offset
> plot(tree,no.margin=TRUE,label.offset=hh,edge.width=2)
> nodelabels(pie=XX$ace,piecol=c("blue","red","green"), cex=0.5)
> tiplabels(pie=to.matrix(x,colnames(XX$ace)), piecol=c("blue","red","green"),cex=0.5)
> packageVersion("phytools")
[1] ‘0.2.54’
>
> # first simulate some data to use in the demo
> Q<-matrix(c(-2,1,1,1,-2,1,1,1,-2),3,3)
> rownames(Q)<-colnames(Q)<-letters[1:3]
> tree<-sim.history(pbtree(n=50,scale=1),Q)
> x<-tree$states
>
> # now the demo
> mtrees<-make.simmap(tree,x,model="ER",nsim=100)
make.simmap is sampling character histories conditioned on the transition matrix
Q =
a b c
a -2.041867 1.020934 1.020934
b 1.020934 -2.041867 1.020934
c 1.020934 1.020934 -2.041867
(estimated using likelihood);
and (mean) root node prior probabilities
pi =
a b c
0.3333333 0.3333333 0.3333333
Done.
> XX<-describe.simmap(mtrees,plot=FALSE)
100 trees with a mapped discrete character with states:
a, b, c
trees have 23.93 changes between states on average
changes are of the following types:
a,b a,c b,a b,c c,a c,b
x->y 2.11 2.91 4.62 5.77 4.94 3.58
mean total time spent in each state is:
a b c total
raw 2.2816521 4.1930936 4.2593925 10.73414
prop 0.2125603 0.3906316 0.3968081 1.00000
> hh<-max(nodeHeights(tree))*0.02 # for label offset
> plot(tree,no.margin=TRUE,label.offset=hh,edge.width=2)
> nodelabels(pie=XX$ace,piecol=c("blue","red","green"), cex=0.5)
> tiplabels(pie=to.matrix(x,colnames(XX$ace)), piecol=c("blue","red","green"),cex=0.5)

Obviously, to adjust the pie-sizes & colors we just change the values of cex & piecol.
That's it.