Error in pic(y, tree) : 'phy' is not rooted and fully dichotomous
phylANOVA is a function that does the simulation-based phylogenetic ANOVA of Garland et al. (1993), but with posthoc tests about the group means - also based on simulation. (Note that this is the same as the "geiger" function phy.anova, but with the posthoc comparison of means added in.)
There's no inherent reason why the function should require a fully bifurcating tree. The reason that it does is because I use pic as a quick way to compute the Brownian motion rate of evolution for simulation, and pic needs a binary tree as input. This bug is easily fixed.
I've updated the code. The link to the new code is here. The following shows a simulation of a tree with multifurcations and data for the phylogenetic ANOVA; a failed analysis with the current version of the code; and a successful analysis with the updated version. Note that there are some neat tricks of simulation thrown in here....
> # load phytools
> require(phytools)
> # simulate tree
> tree<-pbtree(n=50)
> # set some branches to zero to create polytomies
> tree$edge.length[tree$edge.length<0.1]<-0
> is.ultrametric(tree) # check if ultrametric
[1] FALSE
> # now add some length to terminal branches so that
> # the tree is ultrametric
> addTip<-max(vcv(tree))-diag(vcv(tree))
> tree$edge.length[tree$edge[,2]<=length(tree$tip)]<- tree$edge.length[tree$edge[,2]<=length(tree$tip)]+addTip
> tree<-di2multi(tree)
> is.ultrametric(tree) # check if ultrametric
[1] TRUE
> is.binary.tree(tree) # check if bifurcating
[1] FALSE
> # let's take a look at our tree with polytomies
> plotTree(tree,fsize=0.8)
> require(phytools)
> # simulate tree
> tree<-pbtree(n=50)
> # set some branches to zero to create polytomies
> tree$edge.length[tree$edge.length<0.1]<-0
> is.ultrametric(tree) # check if ultrametric
[1] FALSE
> # now add some length to terminal branches so that
> # the tree is ultrametric
> addTip<-max(vcv(tree))-diag(vcv(tree))
> tree$edge.length[tree$edge[,2]<=length(tree$tip)]<- tree$edge.length[tree$edge[,2]<=length(tree$tip)]+addTip
> tree<-di2multi(tree)
> is.ultrametric(tree) # check if ultrametric
[1] TRUE
> is.binary.tree(tree) # check if bifurcating
[1] FALSE
> # let's take a look at our tree with polytomies
> plotTree(tree,fsize=0.8)
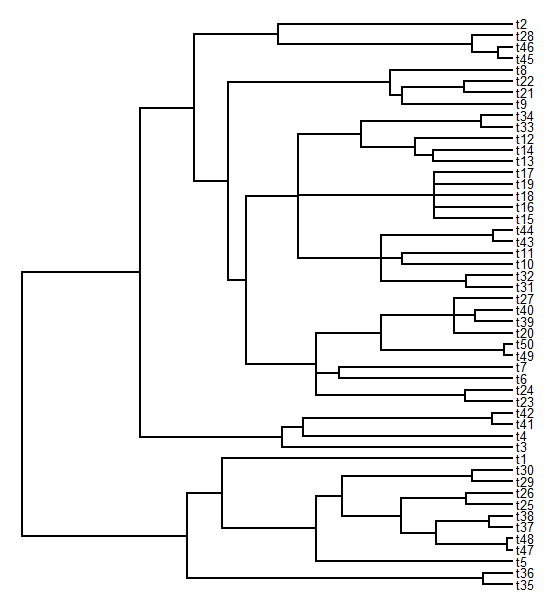
> # ok now let's create a discrete character on the tree
> Q<-matrix(c(-2,1,1,1,-2,1,1,1,-2),3,3)
> rownames(Q)<-colnames(Q)<-c("A","B","C")
> x<-sim.history(tree,Q)$states
> # now let's add an effect that depends on the tip state
> # for the discrete trait
> X<-sapply(c("A","B","C"),"==",x)*1
> y<-X%*%c(0,2,6)+fastBM(tree)
> # conduct phylogenetic ANOVA (current function)
> phylANOVA(tree,x,y) # fails
Error in pic(y, tree) : 'phy' is not rooted and fully dichotomous
> # with a multi2di resolved tree
> phylANOVA(multi2di(tree),x,y)
$F
[1] 10.11326
$Pf
[1] 0.003
$T
A B C
A 0.000000 -1.514359 -4.496845
B 1.514359 0.000000 -2.618537
C 4.496845 2.618537 0.000000
$method
[1] "holm"
$Pt
A B C
A 1.000 0.185 0.003
B 0.185 1.000 0.006
C 0.003 0.006 1.000
> # load updated version
> source("phylANOVA.R")
> phylANOVA(tree,x,y)
$F
[1] 10.11326
$Pf
[1] 0.001
$T
A B C
A 0.000000 -1.514359 -4.496845
B 1.514359 0.000000 -2.618537
C 4.496845 2.618537 0.000000
$method
[1] "holm"
$Pt
A B C
A 1.000 0.172 0.003
B 0.172 1.000 0.014
C 0.003 0.014 1.000
> Q<-matrix(c(-2,1,1,1,-2,1,1,1,-2),3,3)
> rownames(Q)<-colnames(Q)<-c("A","B","C")
> x<-sim.history(tree,Q)$states
> # now let's add an effect that depends on the tip state
> # for the discrete trait
> X<-sapply(c("A","B","C"),"==",x)*1
> y<-X%*%c(0,2,6)+fastBM(tree)
> # conduct phylogenetic ANOVA (current function)
> phylANOVA(tree,x,y) # fails
Error in pic(y, tree) : 'phy' is not rooted and fully dichotomous
> # with a multi2di resolved tree
> phylANOVA(multi2di(tree),x,y)
$F
[1] 10.11326
$Pf
[1] 0.003
$T
A B C
A 0.000000 -1.514359 -4.496845
B 1.514359 0.000000 -2.618537
C 4.496845 2.618537 0.000000
$method
[1] "holm"
$Pt
A B C
A 1.000 0.185 0.003
B 0.185 1.000 0.006
C 0.003 0.006 1.000
> # load updated version
> source("phylANOVA.R")
> phylANOVA(tree,x,y)
$F
[1] 10.11326
$Pf
[1] 0.001
$T
A B C
A 0.000000 -1.514359 -4.496845
B 1.514359 0.000000 -2.618537
C 4.496845 2.618537 0.000000
$method
[1] "holm"
$Pt
A B C
A 1.000 0.172 0.003
B 0.172 1.000 0.014
C 0.003 0.014 1.000
I Liam. I'm getting a error from this code which I can't figure out.
ReplyDeletephanova <- phylANOVA(grantree, dat1$RR, dat1$LOCOM, posthoc=T, p.adj="holm")
Error in if (ssr < 1e-10 * mss) warning("ANOVA F-tests on an essentially perfect fit are unreliable") :
missing value where TRUE/FALSE needed
In addition: Warning messages:
1: In model.response(mf, "numeric") :
using type="numeric" with a factor response will be ignored
2: In Ops.factor(y, z$residuals) : - not meaningful for factors
3: In Ops.factor(object$residuals, 2) : ^ not meaningful for factors
>