A phytools user asked the following question:
"I know I can constrain the state of the root node, but is it possible to also constrain the state at specific internal nodes?"
This is possible, although it requires a hack. We can basically attach a terminal edge (a tip) of zero length to any of the nodes that we want to constrain. We can then assign a values to that tip that is the value to which we want to constrain that node. After we conduct our stochastic mapping, we can then prune all the zero length terminal edges we've added from our stochastically mapped trees.
Here is a quick demo:
> ## first let's simulate a true character history
> tree<-pbtree(n=26,tip.label=LETTERS,scale=1)
> Q<-matrix(c(-2,2,2,-2),2,2)
> rownames(Q)<-colnames(Q)<-letters[1:2]
> true.history<-sim.history(tree,Q)
> cols<-setNames(c("blue","red"),letters[1:2])
> plotSimmap(true.history,cols)
> nodelabels()
> ## now let's run our analysis as normal
> ## (i.e., without constraint)
> x<-getStates(true.history,"tips")
> trees<-make.simmap(tree,x,nsim=100)
make.simmap is sampling character histories conditioned on the transition matrix
Q =
a b
a -2.29596 2.29596
b 2.29596 -2.29596
(estimated using likelihood);
and (mean) root node prior probabilities
pi =
a b
0.5 0.5
Done.
> ## now let's summarize the posterior probabilities
> ## at each node
> plotSimmap(trees[[1]],cols) ## one of our maps
> obj<-describe.simmap(trees)
> nodelabels(pie=obj$ace,piecol=cols,cex=0.6)
> ## next let's constrain, say, node 46 to be
> ## its true value
> x<-c(getStates(true.history,"tips"),
getStates(true.history,"nodes")["46"])
> tt<-bind.tip(tree,"46",edge.length=0,where=46)
> plotTree(tt)
> trees<-make.simmap(tt,x,nsim=100)
make.simmap is sampling character histories conditioned on the transition matrix
Q =
a b
a -2.219295 2.219295
b 2.219295 -2.219295
(estimated using likelihood);
and (mean) root node prior probabilities
pi =
a b
0.5 0.5
Done.
> ## now drop all tips "46" from the trees
> trees<-lapply(trees,drop.tip.simmap,tip="46")
> class(trees)<-"multiPhylo"
> ## now let's once again summarize our results:
> plotSimmap(trees[[1]],cols) ## one of our maps
> obj<-describe.simmap(trees)
> nodelabels(pie=obj$ace,piecol=cols,cex=0.6)
If we want to fix multiple nodes, we have to keep in mind that the node numbering will change every time that we add a tip to the tree.
In the most extreme case we might know the states at each internal node of the tree. In this case, we are just sampling reconstructions of the character history conditioned on the true (known) states at all internal nodes. This is what that might look like:
> ## pull our data vector
> x<-c(getStates(true.history,"tips"),
getStates(true.history,"nodes"))
> nodes<-1:tree$Nnode+length(tree$tip.label)
> for(i in 1:length(nodes)){
if(i==1) tt<-bind.tip(tree,nodes[i],where=nodes[i],edge.length=0)
else {
M<-matchNodes(tree,tt)
tt<-bind.tip(tt,nodes[i],where=M[which(M[,1]==nodes[i]),2],
edge.length=0)
}
}
> plotTree(tt)
> ## now run stochastic mapping
> trees<-make.simmap(tt,x,nsim=100)
make.simmap is sampling character histories conditioned on the transition matrix
Q =
a b
a -2.91958 2.91958
b 2.91958 -2.91958
(estimated using likelihood);
and (mean) root node prior probabilities
pi =
a b
0.5 0.5
Done.
> ## drop all added tips
> trees<-lapply(trees,drop.tip.simmap,
tip=as.character(nodes))
> class(trees)<-"multiPhylo"
> ## visualize the results
> plotSimmap(trees[[1]],cols) ## one of our maps
> obj<-describe.simmap(trees)
> nodelabels(pie=obj$ace,piecol=cols,cex=0.6)
We can even do the following:
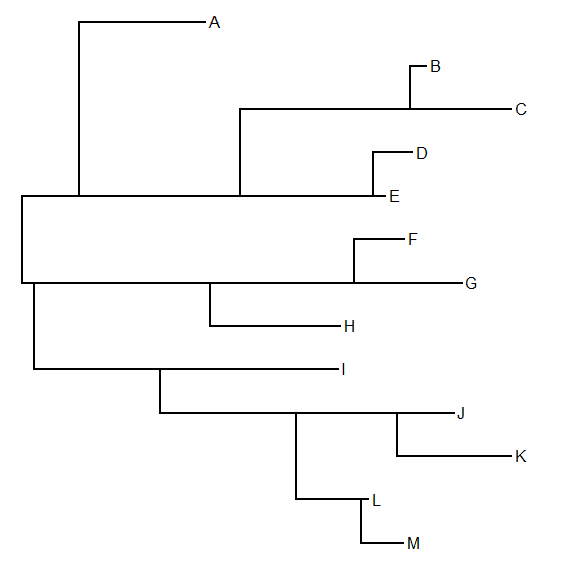
> obj<-densityMap(trees,plot=FALSE)
sorry - this might take a while; please be patient
> plot(obj,lwd=4,outline=TRUE)
Which, in this case, shows the uncertainty about where transitions between states occurred, even when conditioned on knowing the true states at all nodes.
Finally, since make.simmap also allows us to supply a prior on tip states, we can use the same hack to put a prior on internal nodes.
OK. That's it for now.