Today a phytools user emailed with the following question:
"I am still having difficulty running an analysis on the evolution of a continuous character with a partial data set (I have character states for about 50% of the taxa). I would like to display the results using contMap, but since fastAnc is built into the code, it won't map the character because the # of taxa in the data set doesn't match the # of taxa in the tree. I attempted to replace fastAnc with the modified version of anc.ML from one of your blog posts (link), but I couldn't get that to work either. I received the same error message as with fastAnc:
Error in ace(x, a, method = "pic") : length of phenotypic and of phylogenetic data do not match
Is there any way to use contMap with missing data? Any help would be greatly appreciated. Thank you for your time."
They are totally on the right track, but things get a little trickier than they might have anticipated because whereas fastAnc (when used in it's default mode) just returns a vector of estimated character values at nodes, anc.ML (when some taxa are present in the tree but missing from the input data) returns a list with the vector missing.x containing the MLE phenotypic trait values of taxa with missing data. It also happens to be the case that I never got around to adding this updated version of anc.ML to the phytools package.
Well, I have now made both updates so it is possible to (1) specify an inference method (method="fastAnc" or method="anc.ML"); and (2) phytools function anc.ML now permits missing data, in which case the states for the tip taxa in the tree missing from the data vector will be estimated using ML. The code for these updates are here: anc.ML, contMap, but since both use internal functions not in the phytools namespace it is probably wise to download the latest phytools build and install from source.
Here's a quick demo:
[1] ‘0.4.7’
> ## simulate tree & data
> tree<-pbtree(n=26,tip.label=LETTERS)
> x<-fastBM(tree)
> ## compute limits on x to use in both plots
> lims<-c(floor(min(x)*10)/10,ceiling(max(x)*10)/10)
> ## simulate missing tip values
> y<-sample(x,20)
> ## which taxa are missing
> setdiff(tree$tip.label,names(y))
[1] "H" "I" "O" "T" "W" "Z"
> ## contMap from full dataset
> contMap(tree,x,lims=lims,legend=1.5)
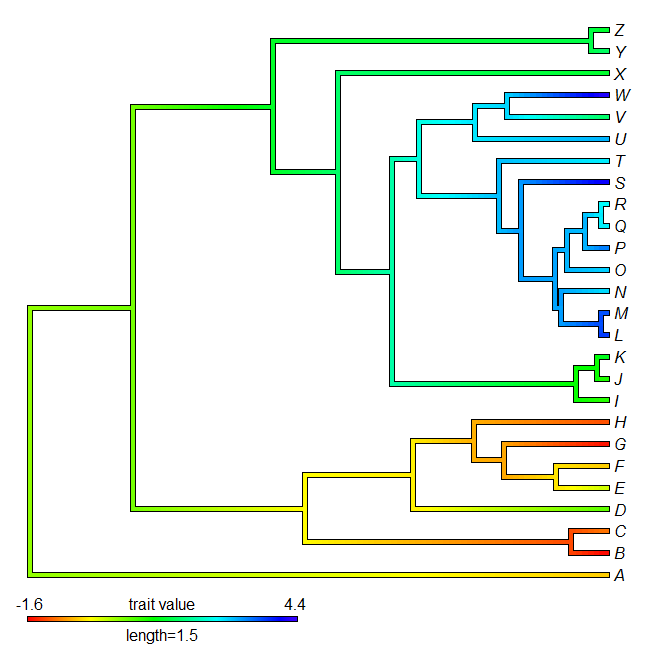
> contMap(tree,y,lims=lims,method="anc.ML",legend=1.5)
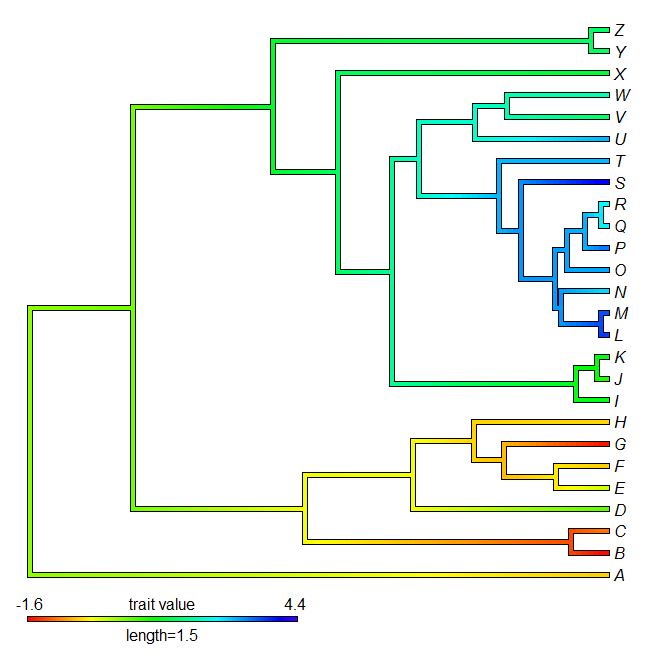
OK - that's it.
Do you think it might be worthwhile to annotate the tips with missing data so which are missing is immediately apparent? Maybe give them a white bubble or something at the very tip?
ReplyDeleteThis can be done easily already by the user because contMap works with ape tiplabels and nodelabels. Good suggestion.
DeleteAmazing work! Beautiful.
ReplyDeleteA general question:
Would it be possible in contMap to specify our own colorramp instead of stuck with the default red-to-blue?
This is not too hard. I just blogged about it here. Let me know if you have any difficulties. - Liam
DeleteHello Dr. Revell,
ReplyDeleteI am very new to R and have been using it for a few weeks. I have a dataset of continuous characters which has missing data. When I used the example above, I could not load the data file(which in .csv format) onto the tree. and also when i ran the code, I got an error which said Error in ace(x, a, method = "pic") :
length of phenotypic and of phylogenetic data do not match.
In addition: Warning message:
In mean.default(x) : argument is not numeric or logical: returning NA.
Also when I use the setdiff function, the missing value taxa are not shown correctly, i.e the ones which have missing values arent shown, instead its random all the time.
Here is the code I ran:
library(ape)
library(phytools)
tree<- read.nexus("data1.nexus")
data<- read.csv("lhpfam.csv")
a<-extract.clade(tree, node=91)
plot(a)
x <- fastBM(a)
col2=2
y<- data
contMap(a,y,method="anc.ML",legend=0.5)
Here is the dataset : https://onedrive.live.com/redir?resid=6d3c647fc5b8119c%21138
(Sorry if this is a duplicate; my other comment didn't go through)
ReplyDeleteI have just started using contMap too! I just got it to work for missing data and here were the pitfalls that I fell into...
(1) Continuous character data should be formatted as row #1 as species names, and row #2 as continuous character data. I think you might need to transpose your data matrix.
(2) If you read in your data file as read.csv, it's read in as a data frame and that throws an error "Error in A[i, 1]/YY[j] : non-numeric argument to binary operator" So I used the unlist() function to convert it into a vector... which seemed to make the function happy!
Overall, here is my code:
tree <- read.tree("Bayes_rooted.tre")
protodata <- read.csv("protodata.csv", header=TRUE)
protovector <- unlist(protodata)
lims<-c(floor(min(protovector)*10)/10,ceiling(max(protovector)*10)/10)
contMap(tree, protovector, lims=lims, legend=1.5, method="anc.ML")
Hopefully that helps and isn't secretly misleading! (Please correct me if I'm wrong!) Thanks Dr. Revell for writing such an awesome function!
Hello Stephanie,
DeleteI did try the code you've posted after transposing my data, but I am still getting the same error .
Hi Liam,
DeleteI also have a tree with more taxa than the data of the continuous variable I want to reconstruct. I was glad to see that there is a possibility to do it using method="anc.ML" in contMap but I couldn't and I got the following error:
Error in ace(x, a, method = "pic") :
NA/NaN/Inf in foreign function call (arg 5)
I really don't understand what's going on. Could you guide me? thank you.
Hi Liam,
ReplyDeleteI also have a tree with more taxa than the data of the continuous variable I want to reconstruct. I was glad to see that there is a possibility to do it using method="anc.ML" in contMap but I couldn't and I got the following error:
Error in ace(x, a, method = "pic") :
NA/NaN/Inf in foreign function call (arg 5)
I really don't understand what's going on. Could you guide me? thank you.
Which symbol can I use for missing data in the csv matrix for discrete characters? Can I use the "?" symbol?
ReplyDeleteDear all, I am new to Phytools and R, and I am trying to reconstruct continuous ancestral states.
ReplyDeleteI could import the tree in Newick format (with branch lenghts, not ultrametric), and imported a csv file containing trait value but with missing data.
RPCS_copy_number<-read.csv("Males.csv",row.names=1)
Then I converted it into a vector
RPCS_copy_number<-as.matrix(RPCS_copy_number)[,1]
But the problem appears when I try to use the FastAnc function:
fit<-fastAnc(mtDNA.tree,RPCS_copy_number,vars=TRUE,CI=TRUE)
It prompts: Error in ace(x, a, method = "pic") :
NA/NaN/Inf in foreign function call (arg 5)
I understand the problem of having missing data in the character matrix is solved, but it seems I am wrong. Can you help me?
I do not know what the problem is -- but here are some things you could try:
Delete1) Resolve polytomies on your tree using multi2di. This shouldn't be the issue, but it is worth checking.
2) Verify that your data are a simple numeric vector with names.
3) Verify that your tree does not have any terminal edge lengths of zero length -- and, in particular, any pairs of taxa with different states for your trait that are separated by zero distance on the reconstructed tree.
4) Verify that you use the argument method="anc.ML" for your contMap.
- Liam
Dear all, I am new to Phytools and R, I also got error fastAnc:
ReplyDeleteError in ace(x, a, method = "pic") : length of phenotypic and of phylogenetic data do not match this. Then I use "anc.ML" method, but unfortunately I got another error: Error in solve.default(C) :
Lapack routine dgesv: system is exactly singular: U[11,11] = 0.
Has anyone encounter this error? What should I do to solve this problem? Thanks so much!
My guess is that this is either because of a mismatch between the taxa in your tree and those in your character vector, or because your character vector does not have names. The easiest way to solve this is probably using the setNames function in base R.
DeleteIf anyone has this error, you can check that your tree is fully resolved. In my case, it's one polytomy of my outgroup at the root that's caused the problem.
DeleteHi. I used all above. But I had this error "length of phenotypic and of phylogenetic data do not match." what to will I do?
ReplyDeletelength of phenotypic and of phylogenetic data do not match.
ReplyDeleteDear Liam,
ReplyDeleteThank you very much for your very efficient instructions.
I managed to get my tree with continuous character mapping and optimization for missing values. However, I have different type of data: missing values and not applicable caractere (e.g. petal size on some species of the tree without petal) where in this case a length optimization is not adequate.
I would just like to colour the branch supporting these few species, with non-applicable values, in white or black rather than a character optimising colour.
I can't seem to find it. Do you think there is a solution in this sense rather than just annotating the tips?
Thank you again for all your work
Here is a solution: http://blog.phytools.org/2023/03/showing-missing-values-as-white-lines.html.
DeleteThank you very much, this is exactly the result I was looking for!
Delete