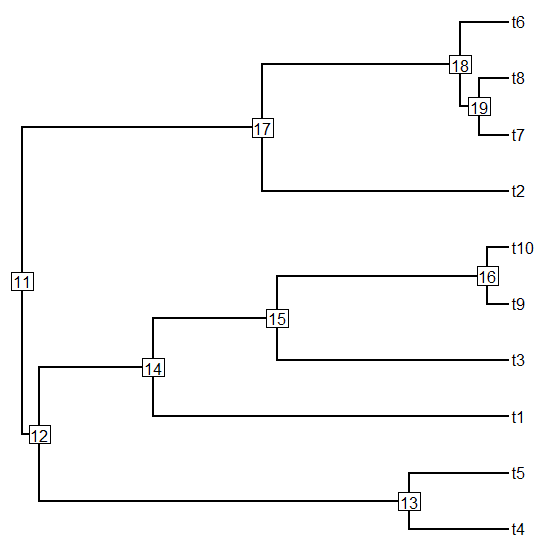I'm working on a manuscript review this Saturday and (I don't think I'm giving away too much about the article to say that) I needed a function to match the node numbers of a tree with the same nodes in a re-rooted version of the
same phylogeny. This differs from the purpose of my previous phytools utility function,
matchNodes, which matches nodes based on the commonality of their descendant tips. This method will not, in general, produce the correct set of matchings for a tree that has merely been rerooted at an internal node.
My idea for obtaining the correct matchings was to use the set of distances to the tip species in the tree. Matching nodes with the same set of patristic distances (or, for trees with arbitrary scale, proportional distances) must be equivalent. Here is the core code of the function:
D1<-dist.nodes(tr1)[1:length(tr1$tip), 1:tr1$Nnode+length(tr1$tip)]
D2<-dist.nodes(tr2)[1:length(tr2$tip), 1:tr2$Nnode+length(tr2$tip)]
rownames(D1)<-tr1$tip.label
rownames(D2)<-tr2$tip.label; D2<-D2[rownames(D1),]
for(i in 1:tr1$Nnode){
z<-apply(D2,2,function(X,y) cor(X,y),y=D1[,i])
Nodes[i,1]<-as.numeric(colnames(D1)[i])
Nodes[i,2]<-as.numeric(names(which(z>=(1-tol))))
}
The first part of the code uses the 'ape' function
dist.nodes to compute the set of distances from each node to all tips; and then sorts them to have the same row-wise ordering in the two matrices. Next, we go through the columns of
D1 and identify the column of
D2 with proportional or equal distances.
Of course - some important bookkeeping has been omitted here. For the full code, see
utilities.R on the phytools page.
Let's try it out:
> tr1<-pbtree(n=20)
> par(mfcol=c(1,2))
> plotTree(tr1,node.numbers=T)
> # re-root tr1 at node=33
> tr2<-root(tr1,node=33)
> plotTree(tr2,node.numbers=T)
> matchNodes(tr1,tr2,method="distances")
tr1 tr2
[1,] 21 NA
[2,] 22 30
[3,] 23 31
[4,] 24 32
[5,] 25 29
[6,] 26 25
[7,] 27 26
[8,] 28 27
[9,] 29 28
[10,] 30 24
[11,] 31 23
[12,] 32 22
[13,] 33 21
[14,] 34 38
[15,] 35 33
[16,] 36 34
[17,] 37 35
[18,] 38 36
[19,] 39 37
> # we can also reverse the order of the trees
> matchNodes(tr2,tr1,method="distances")
tr1 tr2
[1,] 21 33
[2,] 22 32
[3,] 23 31
[4,] 24 30
[5,] 25 26
[6,] 26 27
[7,] 27 28
[8,] 28 29
[9,] 29 25
[10,] 30 22
[11,] 31 23
[12,] 32 24
[13,] 33 35
[14,] 34 36
[15,] 35 37
[16,] 36 38
[17,] 37 39
[18,] 38 34
Careful inspection of the matchings above should (I hope) reveal that the nodes from
tr1 have been correctly matched to the corresponding nodes of
tr2 (in the first example - and the converse in the second case). The column headings in the second example reflect the input order of the trees - not their names in memory.
Note that case 1 leaves one unmatched node - the root. In case two there are no unmatched nodes because all of the
n - 2 nodes in
tr2 have a compatriot in
tr1.
Assuming no major errors are identified, this function will be in the next version of phytools.
That's it.