I just posted code for a new function that conducts the phylogenetic paired t-test of Lindenfors et al. (2010; of which I am also a co-author). Since I am out in the field here on Staniel Cay Exuma, in the Bahamas, it is somewhat difficult for me to error check, etc., so I leave that to my readers. . . . Just kidding, I'm pretty sure there is no major problems with this function, but I am (as always) happy to hear any feedback.
The phylogenetic paired t-test we used in our study was really pretty simple. Basically, we just computed the so-called "phylogenetic mean" for the difference between two values in each species (in our study, these were physiological measures collected from males and females of primates), and we asked if the mean difference was significantly different from zero. In addition, we also incorporated uncertainty about the species means for each trait (essentially following Ives et al. 2007); and we optimized Pagel's λ for the difference.
To use the function, first download it (my R-phylogenetics page; direct link to code here) and load from source; then type:
> result<-phyl.pairedttest(tree,x1,x2)
The result is a list which contains the mean difference, the optimized value of lambda, and some other things.
With a little creativity, this function can also be used to test the hypothesis that the phylogenetic mean for a single trait or a difference between traits is significantly different from an arbitrary value; or to evaluate the phylogenetic signal for a single trait or the difference between two traits.
More soon. . . .
Thursday, May 26, 2011
Sunday, May 22, 2011
Fitting two or more evolutionary correlations to the tree and data
As of late, I am in the field and haven't had time to do much coding (although I am slowly working on implemented the phylogenetic paired t-test of my article with Patrik Lindenfors and Charlie Nunn); however, I thought I would create a quick note about a function that I have available on my R-phylogenetics page, but that I have not described in any earlier blog posts.
The function evol.vcv() fits the model of Revell & Collar (2009). In this article we fit two different BM variance-covariance matrices (we call these "evolutionary rate matrices" - because the diagonal elements are the evolutionary rates, by some account), to different parts of the tree that have been specified a priori by the user. In my implementation (direct link to code here), the user can fit an arbitrary number of traits as well as an arbitrary number of rate matrix categories. These are "painted" on the tree using SIMMAP v1.0 format Newick trees that are read into memory using the function read.simmap() (code here; prior descriptions of the development of this function in several posts).
One of the neat things about this model is that it allows the user to fit different evolutionary correlations between characters to different branches and parts of branches in the tree. For instance, in our study we fit this model for piscivorous and non-piscivorous fish species and found that the evolutionary correlation between two different aspects of buccal morphology was much higher in piscivorous fish species.
To try the function, first load the source:
> source("http://faculty.umb.edu/liam.revell/phytools/evol.vcv/v0.1/evol.vcv.R") # or download
Then, simply read in a data matrix, X, which can contain an arbitrary number of columns, e.g.:
> X<-read.csv(file="data.csv",row.names=1)
Then read the tree in simmap v1.0 format:
> tree<-read.simmap(file="treefile.tre",format="nexus") # for instance
Finally, execute the analysis:
> result<-evol.vcv(tree,X,maxit=5000)
The output is the fit and fitted parameters of both a one matrix and two evolutionary rate matrix model, as well as a comparison of the models.
The function evol.vcv() fits the model of Revell & Collar (2009). In this article we fit two different BM variance-covariance matrices (we call these "evolutionary rate matrices" - because the diagonal elements are the evolutionary rates, by some account), to different parts of the tree that have been specified a priori by the user. In my implementation (direct link to code here), the user can fit an arbitrary number of traits as well as an arbitrary number of rate matrix categories. These are "painted" on the tree using SIMMAP v1.0 format Newick trees that are read into memory using the function read.simmap() (code here; prior descriptions of the development of this function in several posts).
One of the neat things about this model is that it allows the user to fit different evolutionary correlations between characters to different branches and parts of branches in the tree. For instance, in our study we fit this model for piscivorous and non-piscivorous fish species and found that the evolutionary correlation between two different aspects of buccal morphology was much higher in piscivorous fish species.
To try the function, first load the source:
> source("http://faculty.umb.edu/liam.revell/phytools/evol.vcv/v0.1/evol.vcv.R") # or download
Then, simply read in a data matrix, X, which can contain an arbitrary number of columns, e.g.:
> X<-read.csv(file="data.csv",row.names=1)
Then read the tree in simmap v1.0 format:
> tree<-read.simmap(file="treefile.tre",format="nexus") # for instance
Finally, execute the analysis:
> result<-evol.vcv(tree,X,maxit=5000)
The output is the fit and fitted parameters of both a one matrix and two evolutionary rate matrix model, as well as a comparison of the models.
Sunday, May 15, 2011
Field work in the Bahamas
Research in my lab (of one at the moment - but we soon shall be more) takes place in two main areas: computational method development for evolutionary biology; and evolutionary ecology of lizards. The latter calling has me blogging in absentia tonight as I have joined my former Ph.D advisor, Jonathan Losos, and several other colleagues on a 2.5 week field trip to Staniel Cay in the Bahamas. To read more about Jonathan's twenty years (and counting) of field work in the Bahamas, see his blog post here.
Friday, May 13, 2011
New version of optim.phylo.ls()
I just posted a new version of optim.phylo.ls(). This function is for phylogeny inference using the least-squares method. According to this method, we find the tree and branch lengths for which the sum of squared differences between the observed (e.g., measured from DNA sequences) and implied (i.e., the patristic distances on the tree) is minimized. Direct link to code for the new version (v0.4) is here.
My updates arose out of a R-sig-phylo thread on recovering lost branch lengths from a tree when all one has is the topology and a patristic distance matrix. This can be done using the least-squares method (and, in fact, here we are guaranteed to recover the true branch lengths, as noted in Felsenstein 2004). I now make it easy to do so by introducing the option fixed=TRUE which just uses the input tree and stops the inference machinery (which relies on nni() from the {phangorn} package) from running. More on all of this later if I have time.
As an aside - widespread problems with blogger.com means that a post I created yesterday has disappeared! Luckily, I saved a copy (actually, I did this by tracking it down in Google's cache when I first noticed it vanish). I will repost this later if it doesn't reappear on its own.
My updates arose out of a R-sig-phylo thread on recovering lost branch lengths from a tree when all one has is the topology and a patristic distance matrix. This can be done using the least-squares method (and, in fact, here we are guaranteed to recover the true branch lengths, as noted in Felsenstein 2004). I now make it easy to do so by introducing the option fixed=TRUE which just uses the input tree and stops the inference machinery (which relies on nni() from the {phangorn} package) from running. More on all of this later if I have time.
As an aside - widespread problems with blogger.com means that a post I created yesterday has disappeared! Luckily, I saved a copy (actually, I did this by tracking it down in Google's cache when I first noticed it vanish). I will repost this later if it doesn't reappear on its own.
Thursday, May 12, 2011
Issue with extract.clade() relevant to evol.rate.mcmc()
Paolo Piras just clued me into an issue with the {ape} function extract.clade() that affects the pre-processing of the posterior sample from our two-rate Bayesian MCMC analysis performed using evol.rate.mcmc(). Basically, for some orderings of the $edge matrix (in particular, the "cladewise" order given by {phangorn} functions), extract.clade() appears to scramble the tip labels under certain conditions.
For instance, from Paolo's example (but with tip labels altered) if we take the following tree (with nodes labeled according to their indexes in phy$edge):

then extract the clade descended from node "35" and plot it:
> tr<-extract.clade(phy,node=35)
> plot(tr)
we get the following:

Uh-oh - for some reason the tip species "R" & "V" and "T" & "X" have been swapped in the extracted clade!!
It turns out that the easiest way to resolve this is by first computing:
> phy<-read.tree(text=write.tree(phy))
which will guarantee that extract.clade() works properly for this problem. One thing to note, though, is that doing so will change the indexes for the nodes in $edge. Thus, it is essential that this be done before running evol.rate.mcmc() and not after evol.rate.mcmc() but before posterior.evolrate(). I am presently trying to figure out what the smartest way to build this into evol.rate.mcmc() would be.
As an aside, though, this bug is not lethal to evol.rate.mcmc() as the specific ordering of the tip labels in the extracted subtree is not used by posterior.evolrate() (although creating the plotting given here will be affected).
For instance, from Paolo's example (but with tip labels altered) if we take the following tree (with nodes labeled according to their indexes in phy$edge):

then extract the clade descended from node "35" and plot it:
> tr<-extract.clade(phy,node=35)
> plot(tr)
we get the following:

Uh-oh - for some reason the tip species "R" & "V" and "T" & "X" have been swapped in the extracted clade!!
It turns out that the easiest way to resolve this is by first computing:
> phy<-read.tree(text=write.tree(phy))
which will guarantee that extract.clade() works properly for this problem. One thing to note, though, is that doing so will change the indexes for the nodes in $edge. Thus, it is essential that this be done before running evol.rate.mcmc() and not after evol.rate.mcmc() but before posterior.evolrate(). I am presently trying to figure out what the smartest way to build this into evol.rate.mcmc() would be.
As an aside, though, this bug is not lethal to evol.rate.mcmc() as the specific ordering of the tip labels in the extracted subtree is not used by posterior.evolrate() (although creating the plotting given here will be affected).
Tuesday, May 10, 2011
Bug fix in ltt()
One of my Phylogenetics students, Craig Sadowski, just found a bug in the latest version of my function ltt() which computes and creates a lineage-through-time plot; as well as calculating and hypothesis testing the γ statistic of Pybus & Harvey (2000). Basically, when I moved tol (a tolerance value for rounding) out of the function call, I neglected to assign it a value. Oops. Fixed in v0.5 (code here).
Parameter estimates from evol.rate.mcmc()
My coauthors (1,2,3) and I are nearing completion of the revisions for the manuscript describing our MCMC method for identifying the location of a shift in the evolutionary rate through time. I have described the approach and our methods for collapsing the posterior sample in several prior posts (e.g., 1,2,3,4,5,6,7).
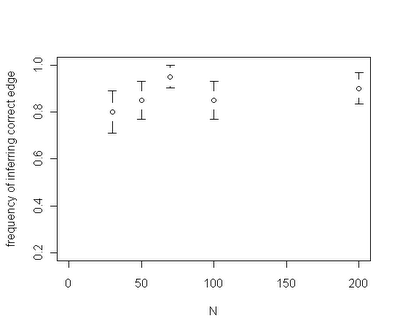
Among the interesting results that have been revealed so far - for a given rate shift our capacity to identify the correct edge on the tree where the rate shift occurred seems to be fairly insensitive to the total size of the tree. For instance, the plot below is for trees of size ranging from N=30 through N=200:
However, we have also found that the ratio of estimated rates (i.e., σ22/σ12) is downwardly biased for small trees, e.g., from an example in which the generating rate ratio σ22/σ12=10:
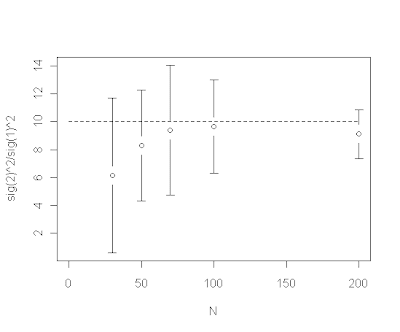
In other words, for small trees the estimated evolutionary rates are too similar relative to the generating rates. We believe, and explain in our manuscript, that this is a direct consequence of integrating across uncertainty in the location of the rate shift (thanks to BR for this insight). For a given rate shift, the posterior density for the location of the shift tends to be much more diffuse on a smaller tree. It makes sense therefore that the estimated difference in evolutionary rates (here, the ratio) will be downwardly biased because we have many posterior samples on incorrect branches where (at least on one side of this split) low and high rates are averaged.
By the way, the plots above were created using plotCI() from the {gplots} package.
More on this and more minor updates to evol.rate.mcmc() (latest version here) coming soon.
Among the interesting results that have been revealed so far - for a given rate shift our capacity to identify the correct edge on the tree where the rate shift occurred seems to be fairly insensitive to the total size of the tree. For instance, the plot below is for trees of size ranging from N=30 through N=200:

However, we have also found that the ratio of estimated rates (i.e., σ22/σ12) is downwardly biased for small trees, e.g., from an example in which the generating rate ratio σ22/σ12=10:

In other words, for small trees the estimated evolutionary rates are too similar relative to the generating rates. We believe, and explain in our manuscript, that this is a direct consequence of integrating across uncertainty in the location of the rate shift (thanks to BR for this insight). For a given rate shift, the posterior density for the location of the shift tends to be much more diffuse on a smaller tree. It makes sense therefore that the estimated difference in evolutionary rates (here, the ratio) will be downwardly biased because we have many posterior samples on incorrect branches where (at least on one side of this split) low and high rates are averaged.
By the way, the plots above were created using plotCI() from the {gplots} package.
More on this and more minor updates to evol.rate.mcmc() (latest version here) coming soon.
Friday, May 6, 2011
CEE symposium online
Dave Bapst pointed out to me the other day that all of the talks from the Centre for Ecology and Evolution's spring symposium, "Integrating Ecology into Macroevolutionary Research," are available online.
Yesterday during lunch I watched a very interesting talk by Gavin Thomas (link here). The photo below is of Gavin talking about my 2008 Systematic Biology paper with Luke Harmon and Dave Collar. (I promise that this is just a minor point in his talk - but I couldn't resist.)

Other talks in the symposium include seminars by Luke Harmon, Andy Purvis, Ally Phillimore, and others.
Yesterday during lunch I watched a very interesting talk by Gavin Thomas (link here). The photo below is of Gavin talking about my 2008 Systematic Biology paper with Luke Harmon and Dave Collar. (I promise that this is just a minor point in his talk - but I couldn't resist.)

Other talks in the symposium include seminars by Luke Harmon, Andy Purvis, Ally Phillimore, and others.
Thursday, May 5, 2011
Discrepant results from fitContinuous(...,method="lambda") and phylosig()
I just received the following email query (edited for brevity):
"I encountered different results of lambda using your phylosig code and geiger's fitContinuous for some of my datasets.... I'm sending one of my datasets and the phylogenetic tree, and below are the codes that I've used to produce the results...."
I downloaded his code and dataset, and, indeed, the results appeared to be different. In particular:
> phylosig(tree,x,method="lambda")
$lambda
[1] 0.9527932
$logL
[1] -255.0452
while:
> fitContinuous(tree,x,model="lambda")
Fitting lambda model:
$Trait1
$Trait1$lnl
[1] -259.0189
$Trait1$beta
[1] 20
$Trait1$lambda
[1] 0.2444363
...
Uhoh. My first thought, naturally, was that the beta version of my phylosig() function has an error (and indeed it still might). But then looking a little more closely at the result of fitContinuous() I started to become a little suspicious. In particular, I noticed that $Trait1$beta (the fitted value of σ2 - the evolutionary rate for this model) was exactly 20. This led me to suspect that fitContinuous() probably does its multivariate optimization using bounds that did not include the MLE of σ2 for these data. If so, this should be resolved by rescaling the data as λ is scale insensitive. Indeed, it did:
> phylosig(tree,x/10,method="lambda")
$lambda
[1] 0.9527932
$logL
[1] -135.3108
> fitContinuous(tree,x/10,model="lambda")
Fitting lambda model:
$Trait1
$Trait1$lnl
[1] -135.3108
$Trait1$beta
[1] 0.661207
$Trait1$lambda
[1] 0.9527761
...
Although it is not clear what bounds are used by default in fitContinuous(), it is straightforward to adjust these. In this case, we would just do:
> fitContinuous(tree,x,model="lambda",bounds=list(beta=c(0,100)))
Fitting lambda model:
$Trait1
$Trait1$lnl
[1] -255.0452
$Trait1$beta
[1] 66.11927
$Trait1$lambda
[1] 0.9527657
...
Code for my phylosig() function is of course available on my website.
"I encountered different results of lambda using your phylosig code and geiger's fitContinuous for some of my datasets.... I'm sending one of my datasets and the phylogenetic tree, and below are the codes that I've used to produce the results...."
I downloaded his code and dataset, and, indeed, the results appeared to be different. In particular:
> phylosig(tree,x,method="lambda")
$lambda
[1] 0.9527932
$logL
[1] -255.0452
while:
> fitContinuous(tree,x,model="lambda")
Fitting lambda model:
$Trait1
$Trait1$lnl
[1] -259.0189
$Trait1$beta
[1] 20
$Trait1$lambda
[1] 0.2444363
...
Uhoh. My first thought, naturally, was that the beta version of my phylosig() function has an error (and indeed it still might). But then looking a little more closely at the result of fitContinuous() I started to become a little suspicious. In particular, I noticed that $Trait1$beta (the fitted value of σ2 - the evolutionary rate for this model) was exactly 20. This led me to suspect that fitContinuous() probably does its multivariate optimization using bounds that did not include the MLE of σ2 for these data. If so, this should be resolved by rescaling the data as λ is scale insensitive. Indeed, it did:
> phylosig(tree,x/10,method="lambda")
$lambda
[1] 0.9527932
$logL
[1] -135.3108
> fitContinuous(tree,x/10,model="lambda")
Fitting lambda model:
$Trait1
$Trait1$lnl
[1] -135.3108
$Trait1$beta
[1] 0.661207
$Trait1$lambda
[1] 0.9527761
...
Although it is not clear what bounds are used by default in fitContinuous(), it is straightforward to adjust these. In this case, we would just do:
> fitContinuous(tree,x,model="lambda",bounds=list(beta=c(0,100)))
Fitting lambda model:
$Trait1
$Trait1$lnl
[1] -255.0452
$Trait1$beta
[1] 66.11927
$Trait1$lambda
[1] 0.9527657
...
Code for my phylosig() function is of course available on my website.
Animation of branching random diffusion
I just created a function to animate branching random diffusion (i.e., Brownian motion with speciation) in discrete time. The functions (optionally) calls several functions from the R package {animation}, and to save the resulting video to file, requires the installation of 'FFmpeg'. My function is available on my R-phylogenetics page (direct link here).
To run it after having installed {animation} and 'FFmpeg', first load the function from source:
> source("branching.diffusion.R")
then, try with the default options (but saving the animation to file using the filename record="diffusion.mp4"):
> branching.diffusion(record="diffusion.mp4")
Be warned, all the animation functions eat up lots of memory. If you just want to view the animation in R without saving it, simply set record=NULL (the default), i.e.:
> branching.diffusion()
The user can also change the variance of the BM process, the birth-rate, and the time of the simulation. One option, smooth, tells the function whether to destroy the existing plot and create a new one each generation using plot() (which results in a slower animation, but is smooth); or add the new bits of evolution using lines() (which results in a faster but choppy animation - although the saved movie should be unaffected). You can also specify the pause between generations (default, 0.02s) and the path to ffmpeg.exe (default path="C:/Program Files/ffmpeg/bin/ffmpeg.exe"). The video below was created using the function on its default settings. Enjoy!
To run it after having installed {animation} and 'FFmpeg', first load the function from source:
> source("branching.diffusion.R")
then, try with the default options (but saving the animation to file using the filename record="diffusion.mp4"):
> branching.diffusion(record="diffusion.mp4")
Be warned, all the animation functions eat up lots of memory. If you just want to view the animation in R without saving it, simply set record=NULL (the default), i.e.:
> branching.diffusion()
The user can also change the variance of the BM process, the birth-rate, and the time of the simulation. One option, smooth, tells the function whether to destroy the existing plot and create a new one each generation using plot() (which results in a slower animation, but is smooth); or add the new bits of evolution using lines() (which results in a faster but choppy animation - although the saved movie should be unaffected). You can also specify the pause between generations (default, 0.02s) and the path to ffmpeg.exe (default path="C:/Program Files/ffmpeg/bin/ffmpeg.exe"). The video below was created using the function on its default settings. Enjoy!
Tuesday, May 3, 2011
Comment on evol.rate.mcmc()
Yesterday a user reported that when he tried to reproduce my example analysis for the function evol.rate.mcmc() here, he encountered the following error when pre-processing the posterior sample:
> mcmc<-posterior.evolrate(tree,minsplit,result$mcmc[201:1001,], result$tips[201:1001])
Error in if (where == 0 || where == "root") where <- ROOTx :
missing value where TRUE/FALSE needed
Today I suggested he try updating {ape} - he did, and the problem went away.
I'm not sure what the compatibility issue is here, but I suspect it has something to do with one of drop.tip(), extract.clade(), or bind.tree(), as these are all called extensively in this function. Take home message - update {ape}!
To find out what version of {ape} (or any other package) you are working with, simply type:
> packageDescription("ape")["Version"]
$Version
[1] "2.7-1"
(FYI 2.7-1 is in fact the version I have installed on this machine.)
> mcmc<-posterior.evolrate(tree,minsplit,result$mcmc[201:1001,], result$tips[201:1001])
Error in if (where == 0 || where == "root") where <- ROOTx :
missing value where TRUE/FALSE needed
Today I suggested he try updating {ape} - he did, and the problem went away.
I'm not sure what the compatibility issue is here, but I suspect it has something to do with one of drop.tip(), extract.clade(), or bind.tree(), as these are all called extensively in this function. Take home message - update {ape}!
To find out what version of {ape} (or any other package) you are working with, simply type:
> packageDescription("ape")["Version"]
$Version
[1] "2.7-1"
(FYI 2.7-1 is in fact the version I have installed on this machine.)
Update to phylosig() - important for large trees
Diego Bilski just emailed me with the following text regarding a problem with my phylosig() function for the estimation of phylogenetic signal:
"With more than about 150 taxa, the lambda calculations of lnlik and p values are not possible, but I wasn't able to identify what causes it. With fewer taxa there are no problems. Even with a simple simulation the same 23 warnings are reported:
> library(ape)
> tree<-rtree(200)
> x<-rTraitCont(tree)
> s.lambda<-phylosig(tree,x,method="lambda",test=TRUE)
There were 23 warnings (use warnings() to see them)"
When I tried to reproduce Diego's error, I could do so easily:
>require(ape)
> source("phylosig.R")
> tree<-rtree(200)
> x<-rTraitCont(tree)
> test<-phylosig(tree,x,method="lambda")
There were 14 warnings (use warnings() to see them)
> test
$lambda
[1] 1.091754
$logL
[1] 87.0783
> warnings()
Warning messages:
1: In optimize(f = likelihood, interval = c(0, maxLambda), ... :
NA/Inf replaced by maximum positive value
2: In ...
I quickly suspected that the problem was with the following calculation in the expression for the log-likelihood:
log(det(sig2*Cl))
This is the calculation of the logarithm of the determinant of the evolutionary rate × the lambda-transformed VCV matrix for the tree. The problem is due to the fact that for moderately large trees det(sig2*vcv(tree) can very easily escape the range of numerical precision of R (i.e., it will become infinitesimal or very large) and thus will be set to 0 or Inf respectively. This will make log(det(...)) Inf or -Inf.
Fortunately, there is a second function to compute the matrix determinant in R, determinant(), and the advantage of this function is that it has an option logarithm, which if set to TRUE returns the logarithm of the determinant. This is perfect for us as we are computing the log-likelihood anyway and this will allow us to compute the log-determinant for matrices in which log(det(...)) will not evaluate.
Indeed, substituting determinant(...,logarithm+TRUE) for log(det(...)) seems to solve our problem from before:
> source("phylosig.R") # updated version
> test<-phylosig(tree,x,method="lambda")
> test
$lambda
[1] 0.9766446
$logL
[1] 156.8490
The new version of phylosig() is now available on my R-phylogenetics page (direct link here).
"With more than about 150 taxa, the lambda calculations of lnlik and p values are not possible, but I wasn't able to identify what causes it. With fewer taxa there are no problems. Even with a simple simulation the same 23 warnings are reported:
> library(ape)
> tree<-rtree(200)
> x<-rTraitCont(tree)
> s.lambda<-phylosig(tree,x,method="lambda",test=TRUE)
There were 23 warnings (use warnings() to see them)"
When I tried to reproduce Diego's error, I could do so easily:
>require(ape)
> source("phylosig.R")
> tree<-rtree(200)
> x<-rTraitCont(tree)
> test<-phylosig(tree,x,method="lambda")
There were 14 warnings (use warnings() to see them)
> test
$lambda
[1] 1.091754
$logL
[1] 87.0783
> warnings()
Warning messages:
1: In optimize(f = likelihood, interval = c(0, maxLambda), ... :
NA/Inf replaced by maximum positive value
2: In ...
I quickly suspected that the problem was with the following calculation in the expression for the log-likelihood:
log(det(sig2*Cl))
This is the calculation of the logarithm of the determinant of the evolutionary rate × the lambda-transformed VCV matrix for the tree. The problem is due to the fact that for moderately large trees det(sig2*vcv(tree) can very easily escape the range of numerical precision of R (i.e., it will become infinitesimal or very large) and thus will be set to 0 or Inf respectively. This will make log(det(...)) Inf or -Inf.
Fortunately, there is a second function to compute the matrix determinant in R, determinant(), and the advantage of this function is that it has an option logarithm, which if set to TRUE returns the logarithm of the determinant. This is perfect for us as we are computing the log-likelihood anyway and this will allow us to compute the log-determinant for matrices in which log(det(...)) will not evaluate.
Indeed, substituting determinant(...,logarithm+TRUE) for log(det(...)) seems to solve our problem from before:
> source("phylosig.R") # updated version
> test<-phylosig(tree,x,method="lambda")
> test
$lambda
[1] 0.9766446
$logL
[1] 156.8490
The new version of phylosig() is now available on my R-phylogenetics page (direct link here).
Bug fix and update to min.split()
I just fixed and updated my function min.split(). This function can be used to analyze the posterior sample of rate shift positions generated by evol.rate.mcmc() (described here: 1,2,3,4,5,6; and in an upcoming paper).
The bug caused the distance between two points on the same edge to be miscalculated. Oops! This should have been the easy part! Any users of evol.rate.mcmc() who have used min.split() should definitely update to v0.5 (here).
The update now allows the user to find the split in a sample that minimizes the summed or the summed squared distances to all the other points in the sample. It can also (optionally) print the distance matrix among splits.
The way the function works is as follows. Take the following set of points on the tree below:

> tree<-read.tree(text="(((1:1.0,2:1.0):1.0,3:2.0):1.0,4:3.0);")
which we represent in a list as follows:
> nodelist<-matrix(c(1,2,7,6,3,4,4,0.5,0.5,0.5,0.5,1.5,1,2), 7,2,dimnames=list(NULL,c("node","bp")))
> nodelist
node bp
[1,] 1 0.5
[2,] 2 0.5
[3,] 7 0.5
[4,] 6 0.5
[5,] 3 1.5
[6,] 4 1.0
[7,] 4 2.0
The syntax here is simple. "node" is the node number in $edge; and "bp" is just the distance along the preceding edge from the node below (not from the "node").
We can then run min.split() as follows:
> source("evol.rate.mcmc.R")
> min.split(tree,nodelist,printD=TRUE)
[,1] [,2] [,3] [,4] [,5] [,6] [,7]
[1,] 0.0 1.0 1.0 2.0 3.0 3.5 4.5
[2,] 1.0 0.0 1.0 2.0 3.0 3.5 4.5
[3,] 1.0 1.0 0.0 1.0 2.0 2.5 3.5
[4,] 2.0 2.0 1.0 0.0 2.0 1.5 2.5
[5,] 3.0 3.0 2.0 2.0 0.0 3.5 4.5
[6,] 3.5 3.5 2.5 1.5 3.5 0.0 1.0
[7,] 4.5 4.5 3.5 2.5 4.5 1.0 0.0
$node
7
$bp
0.5
Which returns the minimum split (in this case node=7,bp=0.5), and, with printD=TRUE, prints to screen the distance matrix among points in the row order of nodelist.
To compute the summed squared distances instead of the summed distances, we just need to do the following:
> min.split(tree,nodelist,method="sumsq")
$node
6
$bp
0.5
and that's it!
The bug caused the distance between two points on the same edge to be miscalculated. Oops! This should have been the easy part! Any users of evol.rate.mcmc() who have used min.split() should definitely update to v0.5 (here).
The update now allows the user to find the split in a sample that minimizes the summed or the summed squared distances to all the other points in the sample. It can also (optionally) print the distance matrix among splits.
The way the function works is as follows. Take the following set of points on the tree below:

> tree<-read.tree(text="(((1:1.0,2:1.0):1.0,3:2.0):1.0,4:3.0);")
which we represent in a list as follows:
> nodelist<-matrix(c(1,2,7,6,3,4,4,0.5,0.5,0.5,0.5,1.5,1,2), 7,2,dimnames=list(NULL,c("node","bp")))
> nodelist
node bp
[1,] 1 0.5
[2,] 2 0.5
[3,] 7 0.5
[4,] 6 0.5
[5,] 3 1.5
[6,] 4 1.0
[7,] 4 2.0
The syntax here is simple. "node" is the node number in $edge; and "bp" is just the distance along the preceding edge from the node below (not from the "node").
We can then run min.split() as follows:
> source("evol.rate.mcmc.R")
> min.split(tree,nodelist,printD=TRUE)
[,1] [,2] [,3] [,4] [,5] [,6] [,7]
[1,] 0.0 1.0 1.0 2.0 3.0 3.5 4.5
[2,] 1.0 0.0 1.0 2.0 3.0 3.5 4.5
[3,] 1.0 1.0 0.0 1.0 2.0 2.5 3.5
[4,] 2.0 2.0 1.0 0.0 2.0 1.5 2.5
[5,] 3.0 3.0 2.0 2.0 0.0 3.5 4.5
[6,] 3.5 3.5 2.5 1.5 3.5 0.0 1.0
[7,] 4.5 4.5 3.5 2.5 4.5 1.0 0.0
$node
7
$bp
0.5
Which returns the minimum split (in this case node=7,bp=0.5), and, with printD=TRUE, prints to screen the distance matrix among points in the row order of nodelist.
To compute the summed squared distances instead of the summed distances, we just need to do the following:
> min.split(tree,nodelist,method="sumsq")
$node
6
$bp
0.5
and that's it!
Monday, May 2, 2011
Plotting the posterior probabilities on the tree (using pies!)
I thought I would post a few words on plotting the posterior probabilities of the rate shift on the tree. This follows specifically from my previous post here.
We can, of course, do this using edgelabels() from {ape} and plot the probabilities in numeric form (boring). We can also use pie graphs! This makes more sense here than for, say, plotting posterior clade probabilities from Bayesian phylogeny inference because in our MCMC method the rate shift must occur somewhere in the tree, so the posterior probabilities of it being anywhere have to sum to 1.0. (Consequently most of the posterior probabilities will often be close to zero and can be left off - which helps us avoid being overwhelms by pies!)
Picking up our simulated example, from before, we can plot the posterior probabilities (for all p>0.01) of the rate shifts on the tree using the following commands:
> p<-hist(mcmc[,"node"], breaks=0.5:(length(tree$tip)+tree$Nnode+0.5))
> plot(tree)
> edgelabels(edge=match(which(p$density>0.01),tree$edge[,2]), pie=p$density[which(p$density>0.01)],piecol=c("black","white"))
Which should produce something close to the tree below:

Note, that we have simulated a very large effect on the evolutionary rate, which explains why the posterior density of the rate shift is so highly concentrated on the correct edge. For empirical data, the posterior density will often be more dispersed across the branches of the tree.
We can, of course, do this using edgelabels() from {ape} and plot the probabilities in numeric form (boring). We can also use pie graphs! This makes more sense here than for, say, plotting posterior clade probabilities from Bayesian phylogeny inference because in our MCMC method the rate shift must occur somewhere in the tree, so the posterior probabilities of it being anywhere have to sum to 1.0. (Consequently most of the posterior probabilities will often be close to zero and can be left off - which helps us avoid being overwhelms by pies!)
Picking up our simulated example, from before, we can plot the posterior probabilities (for all p>0.01) of the rate shifts on the tree using the following commands:
> p<-hist(mcmc[,"node"], breaks=0.5:(length(tree$tip)+tree$Nnode+0.5))
> plot(tree)
> edgelabels(edge=match(which(p$density>0.01),tree$edge[,2]), pie=p$density[which(p$density>0.01)],piecol=c("black","white"))
Which should produce something close to the tree below:

Note, that we have simulated a very large effect on the evolutionary rate, which explains why the posterior density of the rate shift is so highly concentrated on the correct edge. For empirical data, the posterior density will often be more dispersed across the branches of the tree.
Sunday, May 1, 2011
Update to ltt() and calculation of the gamma-statistic
I just updated my lineage-through-time calculation & plotting function ltt(). The updated code is available on my R-phylogenetics page (direct link here). This function does not add much to what is already available with Dan Rabosky's great {laser} package for diversification analyses; but it does allow one to create an LTT plot with extinct tips, which I do not believe is permitted in prior implementations.
I fixed a couple of bugs in this update. The most significant of these created a bug for any tips with zero length. The second, caused an error if the user told the function to drop extinct tips, but in fact all tips in the tree were contemporaneous.
In addition to these bug fixes, I also just added the optional computation of Pybus & Harvey's (2000) γ statistic, along with a two-tailed P-value for this test.
Load and run as follows (with a simulated tree from {geiger}'s birthdeath.tree() as an example):
> source("ltt.R")
> require(geiger)
> tree<-birthdeath.tree(b=1,d=0,taxa.stop=100)
> ltt(tree)
$ltt
101 102 103 112 ...
1 2 3 4 5 ...
$times
101 102 103 112 ...
0.0000000 0.0000000 0.3710933 0.7407788 1.0535915 ...
$gamma
[1] -0.2348588
$p
[1] 0.8143183

I fixed a couple of bugs in this update. The most significant of these created a bug for any tips with zero length. The second, caused an error if the user told the function to drop extinct tips, but in fact all tips in the tree were contemporaneous.
In addition to these bug fixes, I also just added the optional computation of Pybus & Harvey's (2000) γ statistic, along with a two-tailed P-value for this test.
Load and run as follows (with a simulated tree from {geiger}'s birthdeath.tree() as an example):
> source("ltt.R")
> require(geiger)
> tree<-birthdeath.tree(b=1,d=0,taxa.stop=100)
> ltt(tree)
$ltt
101 102 103 112 ...
1 2 3 4 5 ...
$times
101 102 103 112 ...
0.0000000 0.0000000 0.3710933 0.7407788 1.0535915 ...
$gamma
[1] -0.2348588
$p
[1] 0.8143183

Subscribe to:
Posts (Atom)