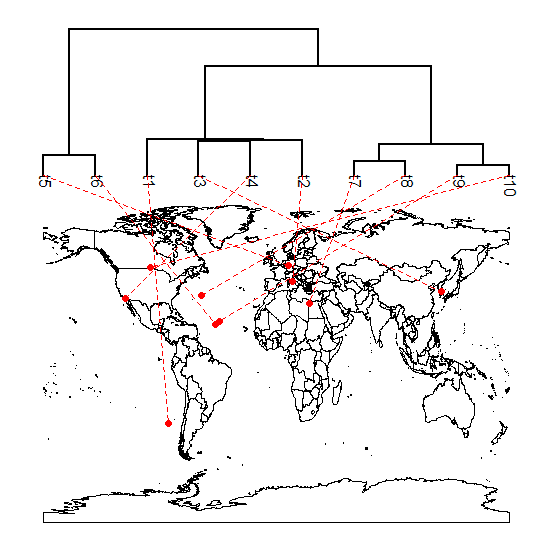
A few days ago I added the option to directly project a phylogeny onto a geographic map into the S3 method plot.phylo.to.map. The most recent phytools version (phytools 0.3-20) is available from my website. It also allows the use of alternative map databases and maps. Here's a quick demo:
> require(phytools)
Loading required package: phytools
> packageVersion("phytools")
[1] ‘0.3.20’
> require(mapdata)
Loading required package: mapdata
> xx<-phylo.to.map(tree,cbind(lat,long), database="worldHires",plot=FALSE)
objective: 286
...
objective: 76
> # first, let's plot the phylogram visualization
> plot(xx,type="phylogram",asp=1.3)
Loading required package: phytools
> packageVersion("phytools")
[1] ‘0.3.20’
> require(mapdata)
Loading required package: mapdata
> xx<-phylo.to.map(tree,cbind(lat,long), database="worldHires",plot=FALSE)
objective: 286
...
objective: 76
> # first, let's plot the phylogram visualization
> plot(xx,type="phylogram",asp=1.3)

> # now let's do a direct projection
> plot(xx,type="direct",asp=1.3)
> plot(xx,type="direct",asp=1.3)
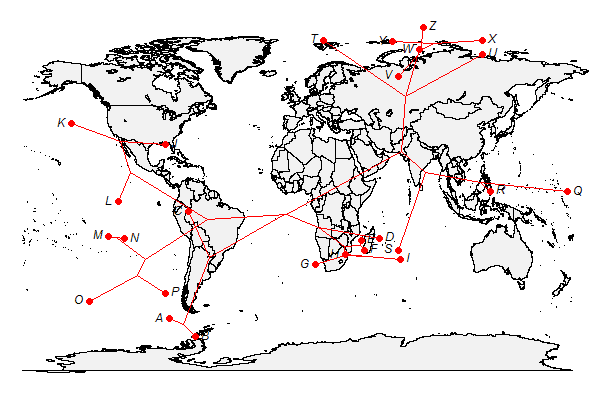
As before, it is a good idea to keep in mind that the nodes in the direct projection do not mean to show ancestral area reconstruction - they are just an attribute of the projection to show relationships among species.