A few days ago I added the option to directly project a phylogeny onto a geographic map into the S3 method plot.phylo.to.map. The most recent phytools version (phytools 0.3-20) is available from my website. It also allows the use of alternative map databases and maps. Here's a quick demo:
> require(phytools)
Loading required package: phytools
> packageVersion("phytools")
[1] ‘0.3.20’
> require(mapdata)
Loading required package: mapdata
> xx<-phylo.to.map(tree,cbind(lat,long), database="worldHires",plot=FALSE)
objective: 286
...
objective: 76
> # first, let's plot the phylogram visualization
> plot(xx,type="phylogram",asp=1.3)
Loading required package: phytools
> packageVersion("phytools")
[1] ‘0.3.20’
> require(mapdata)
Loading required package: mapdata
> xx<-phylo.to.map(tree,cbind(lat,long), database="worldHires",plot=FALSE)
objective: 286
...
objective: 76
> # first, let's plot the phylogram visualization
> plot(xx,type="phylogram",asp=1.3)

> # now let's do a direct projection
> plot(xx,type="direct",asp=1.3)
> plot(xx,type="direct",asp=1.3)
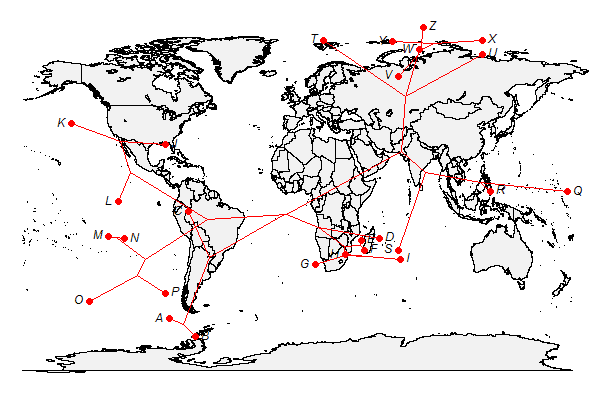
As before, it is a good idea to keep in mind that the nodes in the direct projection do not mean to show ancestral area reconstruction - they are just an attribute of the projection to show relationships among species.
I have not been able to update my version of phytools with the versions that are on this page, What I am doing wrong ..?
ReplyDeleteThe latest version of phytools (not on CRAN) is here: http://www.phytools.org/nonstatic/; however you will need a GCC compiler or Xcode to install from source. If you don't have a compiler installed, and work in Windows, please send me an email and I will build & post a Windows binary.
DeleteI was wondering if there is a way to access aspects of the tree from this function directly, e.g., say I wanted to plot thicker edges or bigger font for tips? Also is there a way to move the lines from the tips down a bit so that the tips are easier to read? thanks!
ReplyDeleteHi Liam, I am trying to run the following command "xx<-phylo.to.map(tree,cbind(lat,long),database="worldHires",plot=FALSE)", but it always returns "tree should be an object of class "phylo"". Could you help me solving this. Great work! Thank you for this great package! Best wishes,
ReplyDeleteIs your object tree of class "phylo"? Find out by running:
Deleteclass(tree)
- Liam
Hi Liam, I am not able to run this with my own data. I checked for typos etc. Does it work for trees with polytomies? Thanks!
ReplyDelete