I just posted a totally rewritten version of the phytools function
make.simmap for stochastic character mapping on trees.
Rich Glor reported that there might be a bug in older versions of
make.simmap because the posterior probabilities at internal nodes were sometimes different than the posterior probabilities computed using Jonathan Bollback's stand-alone Mac OS X program
SIMMAP. That
make.simmap and SIMMAP might give different results was not super concerning to me, because they do slightly different things. Specifically, 1)
make.simmap is meant to sample the root state from the conditional distribution at the root, rather from the conditional distribution × the stationary distribution - as in equation (2) of Bollback (
2006); and 2)
make.simmap is meant to condition on the MLE of
Q, the instantaneous transition matrix, rather than sampling transition rates among states from a user-specified prior distribution. [I believe I'm right about these two aspects of SIMMAP; please correct me if I have any details wrong.]
Anyway, Rich's email inspired me to take a close look at the code, which I'd written originally in mid-2011 and had been little modified since. I did find one bug - specifically, in computing probabilities to sample states from the multinomial, I had rescaled (incorrectly) by the maximum, rather than the sum, of the product of the conditional likelihoods × the transition probabilities - equation (3) in Bollback (
2006). I.e.:
node.states[j,2]<-rstate(p/max(p))
in older versions of
make.simmap (e.g.,
here), should have been:
node.states[j,2]<-rstate(p/sum(p))
This would not have mattered at all if I'd been smart and used the base function
rmultinom, which automatically rescales probabilities internally to sum to 1.0 - but instead I used a custom phytools function
rstate which (aside from also being computationally less efficient), did not. (
rstate still
exists, but now it calls
rmultinom internally.)
The significance of this bug will depend on the specific case. In some empirical datasets that I ran, the effect on the posterior density (computed using
densityMap) was small - but this should probably not be assumed.
In going closely over the code of
make.simmap I also realized it was due a major overhaul. This is hardly surprising as I wrote the original version of this function "way back" in mid-2011 when I was only really starting to become adventurous in R after switching over from doing mostly everything in C. In re-writing
make.simmap I've 'encapsulated' (so to speak) all the different steps of the mapping process into different internally called functions. I also removed the dependency of
make.simmap on the 'ape' function
ace for computing the conditional likelihoods - although in this case by borrowing liberally from the
ace code (with attribution, of course) in a different internally called function,
apeAce.
I also took advantage of the re-write to modify deviation 1) from Bollback's
SIMMAP. Now the user has the option of assuming equal state frequencies, inputting arbitrary state frequencies,
or estimating frequencies by numerically solving for the stationary distribution conditioned on
Q. By multiplying our conditional likelihoods at the root by the stationary distribution, we are basically putting a prior on the root state that is equal to the stationary distribution implied by the fitted model. I'm not totally sure whether we want to do this or not. Feedback welcome.
One interesting case of user-defined state frequencies is the practice of setting the frequency at 1.0 for one state and 0.0 for the others. What this will result in is a set of stochastic maps conditioning on a particular value (the one with probability 1.0) at the root node of the tree - which might be interesting for some problems.
For
make.simmap(...,message=TRUE) (the default), the function will now also spit back the value for the transition matrix,
Q and the prior on the state frequencies at the root (
π) used during simulation.
Finally, this updated version of
make.simmap runs about 30-40% faster than the prior version (even with the specific bug-fix identified above), which is not trivial.
I also built a new version of phytools,
phytools 0.2-27, which can be downloaded and installed from source in the
usual way.
Here's a quick demo of use of
make.simmap. [Note that the input matrix
Q for simulation in
sim.history is the transpose of the printed matrix
Q in
make.simmap. In the literature on molecular evolution models and Markov chains in general there is some discrepancy about whether the rows or columns should sum to 0.0 by convention (the only difference being whether we need to post- or pre-multiply
eQt, respectively, by π
(t) to get π
(t+1)).]
> require(phytools)
> packageVersion("phytools")
[1] ‘0.2.27’
> tree<-pbtree(n=200,scale=1)
> Q<-matrix(c(-1,1,1,-1),2,2)
> rownames(Q)<-colnames(Q)<-letters[1:nrow(Q)]
> tree<-sim.history(tree,Q,anc="a")
> mtrees<-make.simmap(tree,tree$states,model="ER", nsim=100,pi="estimated")
make.simmap is sampling character histories conditioned on the transition matrix
Q =
a b
a -0.7876848 0.7876848
b 0.7876848 -0.7876848
(estimated using likelihood);
and state frequencies (used to sample the root node following Bollback 2006)
pi =
a b
0.5 0.5
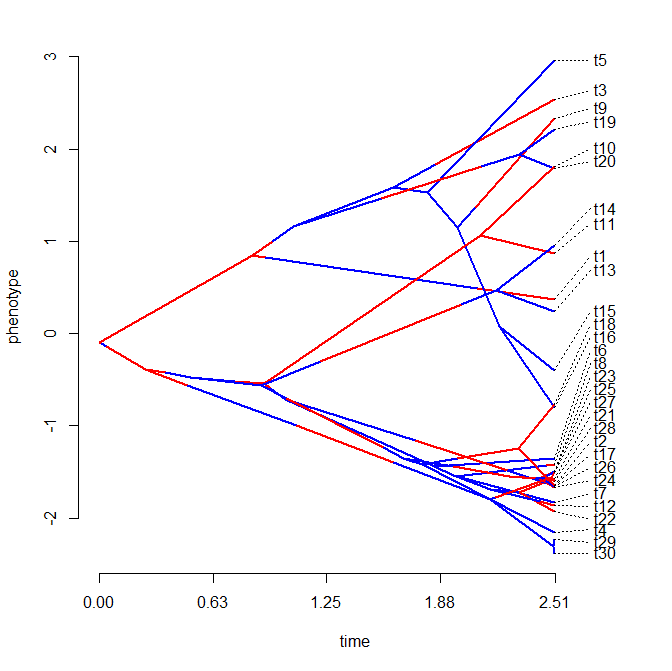
> cols<-setNames(c("blue","red"),letters[1:nrow(Q)])
> plotSimmap(mtrees[[1]],cols,pts=F,ftype="off")
Now let's compute the state frequencies from the stochastic maps for each internal node. These are our posterior probabilities for nodes:
> # function to compute the states
> foo<-function(x){
+ y<-sapply(x$maps,function(x) names(x)[1])
+ names(y)<-x$edge[,1]
+ y<-y[as.character(length(x$tip)+1:x$Nnode)]
+ return(y)
+ }
>
> AA<-sapply(mtrees,foo)
> piesA<-t(apply(AA,1,function(x,levels,Nsim) summary(factor(x,levels))/Nsim,levels=letters[1:3], Nsim=100))
> plot(tree,no.margin=TRUE,show.tip.label=F)
> nodelabels(pie=piesA,cex=0.6,piecol=cols)
Finally, let's compare to the generating map:
> # first compute the number of changes of each type
> XX<-countSimmap(mtrees,message=FALSE)
> colMeans(XX)
N a,a a,b b,a b,b
28.65 0.00 10.88 17.77 0.00
> # compare to the true history
> countSimmap(tree,message=FALSE)
$N
[1] 27
$Tr
a b
a 0 11
b 16 0
> # now compute the mean overlap with the true history
> xx<-sapply(mtrees,map.overlap,tree)
> mean(xx)
[1] 0.9144295
I think that we can be pretty confident that
make.simmap is doing what it's supposed to do; however please keep in mind that this version is brand new and has not been thoroughly tested. And, as Rich showed in pointing out this potential error to me, it is always wise to look behind the curtain - if possible - and don't hesitate to report any bugs, issues, or questionable results to me ASAP. I'll do whatever I can to address them.