Someone recently emailed me about slicing a tree at a particular height from the root and then keeping all the subtrees. This seemed pretty easy, so I thought I would implement it using the {ape} function
extract.clade(). This is how I did it. First, I reordered the tree
"cladewise", using
reorder.phylo(); next, I computed the heights of all the internal and terminal nodes in the tree and put these values into a matrix to match the
$edge matrix from my
"phylo". Then, I found all the edges intersected by my slice and identified the tipward nodes on those edges, excluding all the nodes that were also tips. Finally, I extracted each subtree descended from the nodes using
extract.clade() and added a
$root.edge to each tree depending on where its root branch was sliced. I then returned all of these trees as a
"multiPhylo" object.
The function is posted to my website,
here. Please be warned that I'm not sure what will happen if the slice point is greater than the tree length or if it only intersects terminal edges.
First, let's create a random tree:
> require(geiger)
> tree<-birthdeath.tree(b=1,d=0,taxa.stop=20)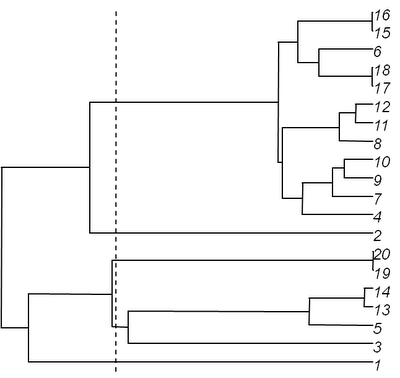
Now, let's slice it at
slice=0.6, shown as the dotted line above (having verified, of course, that this will indeed produce some non-trivial subtrees):
> source("treeSlice.R")
> trees<-treeSlice(tree,0.6)
> plot(trees,root.edge=T)which, in this case, produces the following set of three trees:
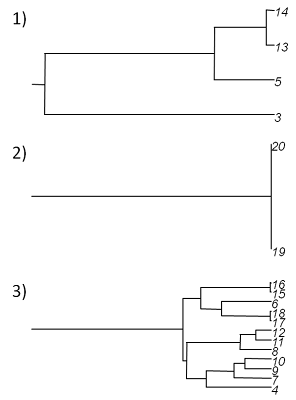
We're done!


Note that this is *not* in phytools 0.0-3.
ReplyDeleteCool!
ReplyDeleteIf it comes up, I've written code for doing the opposite: taking a slice and obtaining the tree up to that slice.
Hi Liam,
ReplyDeleteThanks a lot. This is really helpful function and I am going to use it very often from now on.
best
Rama
@Rama (and others)
ReplyDeleteCan you share how you will be using this function? I have some ideas but wanted to hear other ideas.
Laurence
A neat function, will be using it now to cut a tree of sequence motifs. I'm wondering though whether it's also possible to make sure that single-tip trees are also returned?
ReplyDeleteThanks!
Mikhail
Done. I will post about this shortly.
ReplyDeleteThis comment has been removed by the author.
ReplyDeleteThis comment has been removed by the author.
ReplyDeleteThank you very much for this post. It works.
ReplyDeleteHi I tried your tree slicing program and so far it is giving me different answers. My tree has 255 tips and when I do this:
ReplyDeletetrees<-treeSlice(nj_tree_root,0.07)
I get only one subtree:
str(trees)
Class "multiPhylo"
List of 1
$ :List of 5
..$ edge : int [1:4, 1:2] 4 5 5 4 5 1 2 3
..$ edge.length: num [1:4] 0.0358 0.0569 0.0485 0.0163
..$ tip.label : chr [1:3] "gene1" "gene2" "gene3"
..$ Nnode : int 2
..$ root.edge : num 0.0393
..- attr(*, "class")= chr "phylo"
..- attr(*, "order")= chr "cladewise"
I am not sure what happens to the rest of the tree. I thought this program will slice the tree at a specific height as shown in the example above and then return different parts of the sliced trees.
Also, when I tried this:trees<-treeSlice(nj_tree_root,0.007)
I got 31 trees but the sum of all of these tree tip.labels is way more than total tip.labels in the original tree. Please comment. I can send you all the info you need of my tree.