A R-sig-phylo subscriber recently posted a request as follows:
“[R-sig-phylo] Collapse a clade by tip labels while maintaining
phylogenetic position
branchlizard . Mon, 12 Sep 2016 12:46:56 -0700
I have posted this question at Stack Overflow. I hope this doesn't violate any community rules about double posting.
I probably could have worded the title better, but I am wanting to collapse any clade within a phylogenetic tree (even if the clade has one member) which has a tip label of "foo” and then count the number of tips which were dropped from that specific clade and create a branch with a tip label displaying 35 foos.
The counting portion is easy; however, when I use
drop.tip(rooted.tree,tip=which(rooted.tree$tip.label=='foo'),subtree=TRUE)
the dropped tips do not maintain their position in the tree. Rather, they are all grouped at the end (counted properly however). Is there anyway to collapse a clade by tip labels and maintain its position"
When asked to clarify, the poster responded that what he or she wants to do is convert a tree like this:
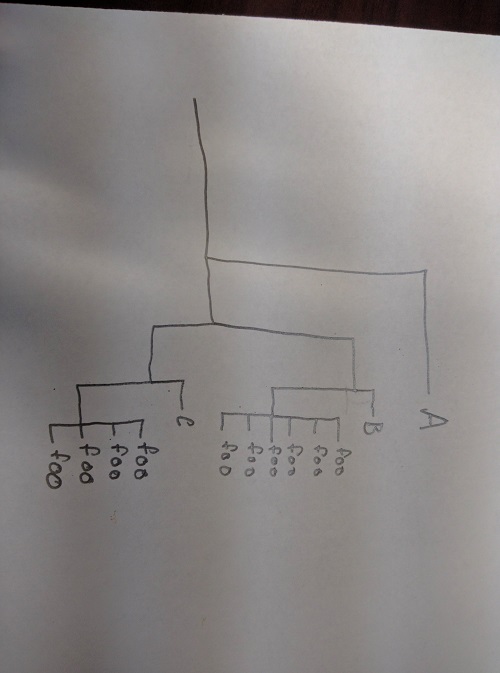
into a tree like this:
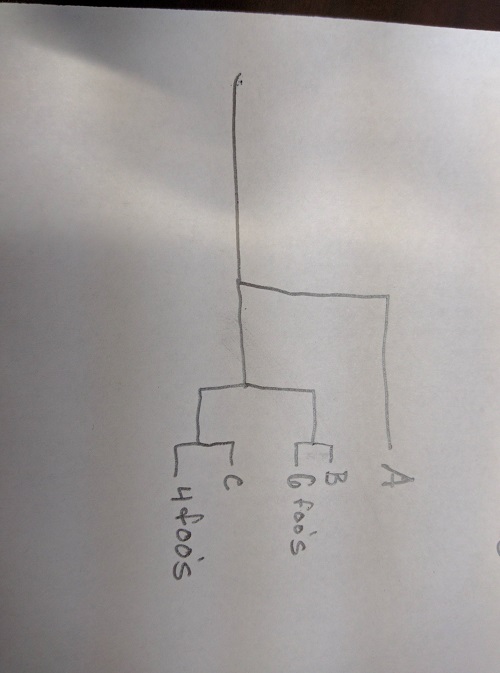
Florian Boucher posted a solution that probably works, I haven't tried. Here is another one:
First load packages and make a tree with this property:
library(phytools)
library(phangorn)
text<-"(((((foo:0.9,foo:0.7):0.7,((foo:0.6,foo:0.9):0.9,foo:0.8):0.5):0.9,A:0.8):0.9,(B:0.1,C:0.1):0.5):0.6,((((foo:0.7,(foo:0.4,foo:0.8):0.9):0.3,(foo:0.9,(foo:0.7,foo:0.2):0.5):0.6):0.7,D:0.7):0.4,((foo:0.3,((((foo:0.3,foo:0.5):0.1,foo:0.5):0.4,foo:0.7):0.3,E:0.1):0.6):1,((F:0.2,G:0.8):0.7,((foo:0.7,foo:0.3):0.2,foo:0.7):0.1):0.8):0.9):0.8);"
tree<-read.tree(text=text)
plotTree(tree)

This is our uncollapsed tree. Now let's go about identifying all the nodes that we want to collapse and collapsing them. One complication is that each time we collapse one clade, the node numbers will, of course, change. A further wrinkle is that because tip labels repeat, it might be difficult to match tips and nodes in the tipical way. Finally, we might want to maintain the tip a certain height about the root - for instance the mean height of the 'foo's of its clade:
nodes<-1:tree$Nnode+Ntip(tree) ## all nodes
subtrees<-list()
for(i in 1:tree$Nnode) subtrees[[i]]<-extract.clade(tree,nodes[i])
names(subtrees)<-nodes ## all subtrees
## all nodes with only "foo" as descendant
all.foos<-nodes[sapply(subtrees,function(x) all(x$tip.label=="foo"))]
foo.mrcas<-all.foos[sapply(all.foos,function(x,tree,y)
!Ancestors(tree,x,"parent")%in%y,
tree=tree,y=all.foos)]
w.foos<-which(tree$tip.label=="foo")
## rename tips uniquely
tree$tip.label[w.foos]<-paste(tree$tip.labe[w.foos],
replicate(length(w.foos),paste(sample(letters,6),
collapse="")),sep="_")
collapsed<-tree
## iterate over all MRCAs of foo clades
for(i in 1:length(foo.mrcas)){
M<-matchNodes(tree,collapsed)
nn<-M[which(M[,1]==foo.mrcas[i]),2]
dd<-Descendants(collapsed,nn)[[1]]
h<-sapply(dd,nodeheight,tree=collapsed)
collapsed$tip.label[dd[1]]<-paste(length(dd),"foo(s)")
ind<-which(collapsed$edge[,2]==dd[1])
collapsed$edge.length[ind]<-collapsed$edge.length[ind]+
mean(h)-h[1]
if(length(dd)>1)
collapsed<-drop.tip(collapsed,
collapsed$tip.label[dd[2:length(dd)]])
}
## finally, address 'singleton' foos, if they exist
ind<-grep("foo_",collapsed$tip.label)
if(length(ind)>0) collapsed$tip.label[ind]<-"1 foo(s)"
Now, let's check our tree:
plotTree(collapsed)

voila!
Hi Liam! Great piece of code! I have been modifying it to collapse clades for non-monophyletic genera on a large tree I'm working with (>2000 tips).
ReplyDeleteIs there any way you can think of to speed up the last loop? matchNodes() seem to take a far bit of time in for each case. At the moment with the tree I'm working with it will take 2 1/2 hours to run.