The most common error reported by users of contMap & densityMap is the following:
Error in while (x > trans[i]) { : missing value where TRUE/FALSE needed
This is usually (but not always) due to the tree having internal edges of zero length, and is just the result of how I set up the internal machinery of these functions. So, for instance:
> tree<-rtree(10)
> tree$edge.length[which(tree$edge[,2]==14)]<-0
> plotTree(tree)
> x<-fastBM(tree)
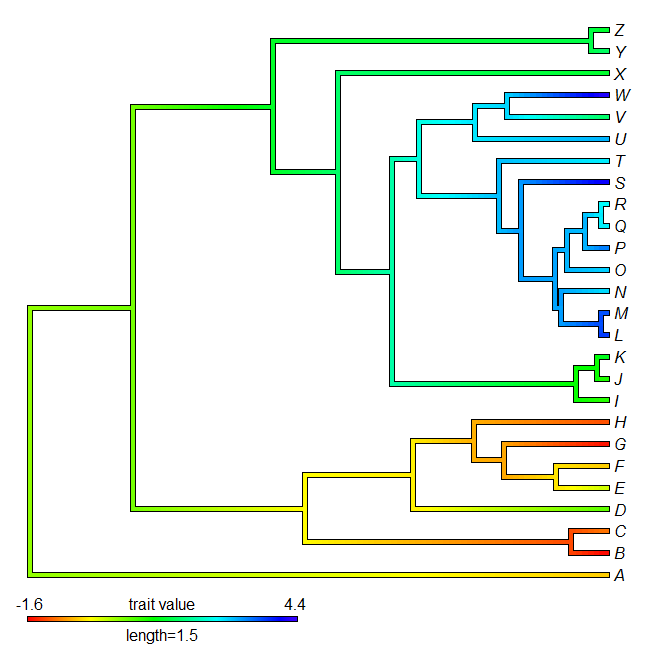
> obj<-contMap(tree,x)
Error in while (x > trans[i]) { : missing value where TRUE/FALSE needed
Obviously, this could be circumvented by first collapsing zero-length branches into polytomies, and this is typically what I have advised users to do - but there is no really good reason why contMap shouldn't be able to work with the zero-length branches still intact. I have just posted a new version with the bug fixed, and it is in a new release of phytools, which can be downloaded & installed from source here.
Here's a demo using the same tree as before:
> packageVersion("phytools")
[1] ‘0.4.10’
> obj<-contMap(tree,x)
I have also added a new function to automate the process of changing the color ramp, described here. This function is called setMap. Here's a demo:
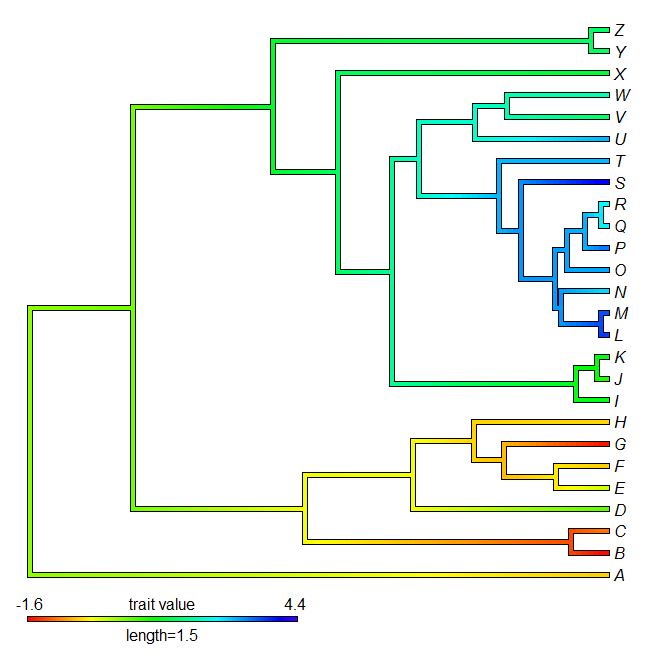
> obj<-setMap(obj,colors=c("blue","green","yellow"), space="Lab")
> plot(obj)
Obviously, there is no way that R can make you use a reasonable color scheme here!
In addition to fixing the same branches of zero-length problem in densityMap, it also now can plot the posterior density of any two-state character, not just a binary character with states of "0" & "1". This required a few small modifications, including accommodating the possibility that someone might use one & only one of states "0" & "1" (and then some other state not "0" or "1"). Here's a demo:
> tree<-pbtree(n=26,tip.label=letters[26:1],scale=1)
> Q<-matrix(c(-1,1,1,-1),2,2)
> rownames(Q)<-colnames(Q)<-LETTERS[1:2]
> x<-sim.history(tree,Q)$states
> trees<-make.simmap(tree,x,nsim=100)
make.simmap is sampling character histories conditioned on the transition matrix
Q =
A B
A -0.5933734 0.5933734
B 0.5933734 -0.5933734
(estimated using likelihood);
and (mean) root node prior probabilities
pi =
A B
0.5 0.5
Done.
> obj<-densityMap(trees,plot=FALSE,res=500)
sorry - this might take a while; please be patient
> plot(obj,outline=TRUE,lwd=6)
That's it for now.