OK, first let's simulate a pure-birth phylogeny for (say) 200 species. We can also simulate our so-called liabilities up the tree with a correlation of 0.8:
> # load phytools & source
> library(phytools)
> source("threshBayes.R")
> # simulate
> tree<-pbtree(n=200,scale=1) # tree
> Xl<-sim.corrs(tree,vcv=matrix(c(1,0.8*sqrt(2),0.8*sqrt(2),2), 2,2)) # liabilities
Now, we can substitute one of these characters (say, trait 1) for a binary trait evolved under the threshold model, setting the threshold to +-0.
> X<-Xl
> X[Xl[,1]<0,1]<-0
> X[Xl[,1]>0,1]<-1
> X
[,1] [,2]
t1 0 -0.05615011
t2 1 0.42579821
t3 1 -0.32314924
t4 0 . . .
Now we are ready to run our MCMC. We'll just use the default control parameters and priors. Note that this may take some time - certainly at least several minutes even on a fast computer.
> mcmc<-threshBayes(tree,X,types=c("discrete","continuous"), ngen=500000)
[1] "gen 1000"
[1] "gen 2000"
[1] "gen 3000"
[1] . . .
The object returned by threshBayes is a list containing the posterior sample for the parameters of the evolutionary model and the liabilities at the tips. Note that for a continuous character liabilities are not sampled (they are treated as known), but these are still given in the posterior sample matrix. Also - we fix the rate of evolution at 1.0 for the liability of our discrete character. This is because don't actually have any information about the scale of our underlying liabilities, and the probability of switching back and forth between states on either side of the threshold is independent of the Brownian rate of evolution of liability (this is probably better explained in Felsenstein 2012).
We can, say, plot the likelihood profile:
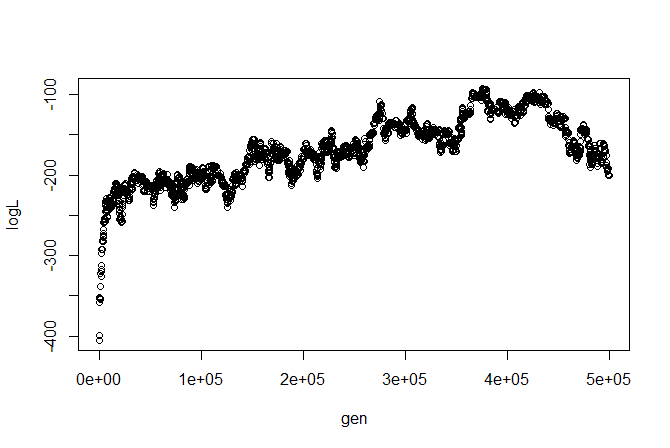
Let's cut off the first half of our posterior sample and get parameter estimates from the second 250,000 generations (2500 samples, in this case):
> estimates<-colMeans(mcmc$par[2501:nrow(mcmc$par),2:6])
> estimates
sig1 sig2 a1 a2 r
1.0000000 2.0309112 0.6746904 1.1889742 0.8341032
These are really close to the generating parameters (particularly for σ2 and r). To be fair, this was part of my motivation in using a "large" tree of 200 taxa, even though run time is considerably longer. Parameter estimates from the posterior sample were not nearly as close on smaller trees.
We are probably most interested in the correlation coefficient - so let's look at the posterior sample for this parameter, with the generating value (0.8, in this case), overlain as a vertical dashed line:
> h<-hist(mcmc$par[2501:nrow(mcmc$par),"r"],0:40/40+0.0125, main=NULL,ylab="freq",xlab="r")
> lines(c(0.8,0.8),c(0,max(h$counts)),lty="dashed")
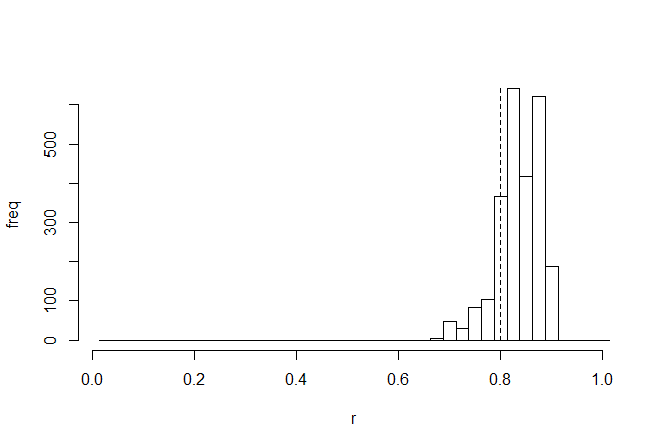
> estLiab<-matrix(colMeans(mcmc$liab[2501:nrow(mcmc$liab), 1:200+1]),200,2)
> cor(Xl[,1],estLiab[,1])
[1] 0.9255481
> plot(Xl[,1],estLiab[,1],xlab="true liability",ylab="estimated liability")

Next up - multiple states for the discrete character!
No comments:
Post a Comment
Note: due to the very large amount of spam, all comments are now automatically submitted for moderation.